Introductions
Maq is a software that builds mapping assemblies from short reads generated by the next-generation sequencing machines. It is particularly designed for Illumina-Solexa 1G Genetic Analyzer, and has preliminary functions to handle ABI SOLiD data.
Maq first aligns reads to reference sequences and then calls the consensus. At the mapping stage, maq performs ungapped alignment. For single-end reads, maq is able to find all hits with up to 2 or 3 mismatches, depending on a command-line option; for paired-end reads, it always finds all paired hits with one of the two reads containing up to 1 mismatch. At the assembling stage, maq calls the consensus based on a statistical model. It calls the base which maximizes the posterior probability and calculates a phred quality at each position along the consensus. Heterozygotes are also called in this process.
Get Maq
Maq is distributed under GNU Public License (GPL). All its source codes are freely available to both academic and commercial users. The latest version can be downloaded at the SourceForge download page. Source codes are also available at the SourceForge subversion server, which can be accessed with:
svn co https://mapass.sourceforge.net/svnroot/mapass/trunk/maq maq
Install Maq
There are two ways to compile maq. The first way is to use the GNU building systems. Simply type:
./configure; make; make installyou can compile and install maq. Three executables, `maq', `maq.pl' and `farm-run.pl', will be copied to /usr/local/bin by default.
Alternatively, you could compile with:
make -f Makefile.genericand manually copy the three executables to the destination directory. Modification to `Makefile.generic' is sometimes needed on different architectures.
Maq is better compiled and used on a 64bit machine because it frequently uses 64bit integers. Compiling as a 32bit executable should work but the speed will be affected.
Get Started with Maq
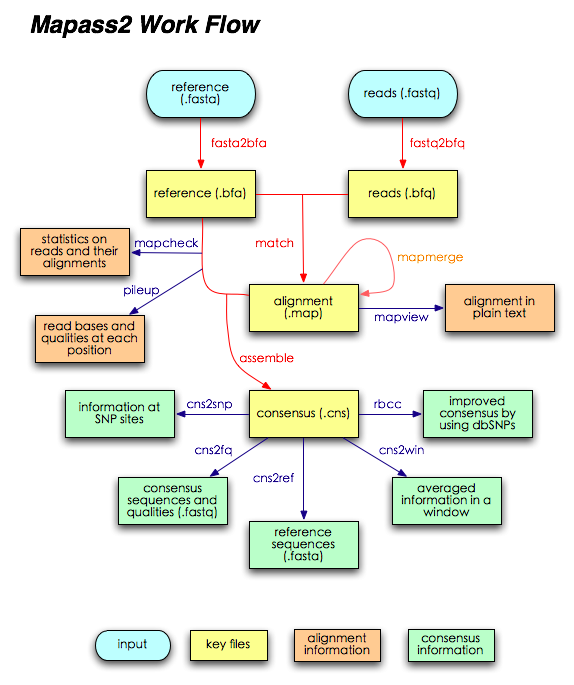
Maq Workflow
IMPORTANT NOTE:
The raw reads format used by Solexa (those `s_?_sequence.txt' from the Solexa pipeline) are different from mapass' FASTQ format in that the qualties are scaled differently. To use maq, you need to first convert the format with:
maq sol2sanger s_1_sequence.txt s_1_sequence.fastqwhere s_1_sequence.txt is the Solexa read sequence file. Missing this step will lead to unreliable SNP calling.
Easyrun Script: maq.pl
For small datasets, the easiest way to run maq is to use the easyrun command of the maq.pl script. This command calls the maq executable and does most of things in batch. If we have a reference ref.fasta and one lane of reads reads.fastq, easyrun can be simply invoked as:
maq.pl easyrun -d outdir ref.fasta reads.fastqwhere ref.fasta is the reference sequence in FASTA format and reads.fastq is the read sequnces in the FASTQ format. Several read files can be applied at the same time on the command line. The results will be put in the outdir directory. For a bacteria genome with several mega bases, the whole process usually takes a few minutes.
Easyrun Output
Several files will be generated by the easyrun command. Endusers may be interested in these ones:
| cns.fq | Consensus sequences and their qualities |
| cns.snp | List of SNPs (format described in the reference manual). |
| assemble.log | Statistics about the coverage and SNPs |
Behind Easyrun
The easyrun command actually executes the following commands in order (see also the workflow chart):
- maq fasta2bfa ref.fasta ref.bfa
- Convert the reference sequences to the binary fasta format
- maq fastq2bfq reads.fastq reads-1.bfq
- Convert the reads to the binary fastq format
- maq match reads-1.map ref.bfa reads-1.bfq
- Align the reads to the reference
- maq mapcheck ref.bfa reads-1.map >mapcheck.txt
- Statistics from the alignment
- maq assemble consensus.cns ref.bfa reads-1.map 2>assemble.log
- Build the mapping assembly
- maq cns2fq consensus.cns >cns.fq
- Extract consensus sequences and qualities
- maq cns2snp consensus.cns >cns.snp
- Extract list of SNPs
Use Maq
To accelerate file I/O and to reduce the use of diskspace, maq first converts reference sequences to the binary fasta (BFA) format and converts reads to binary fastq (BFQ) format. It then aligns reads and builds the mapping assemblies. Most of the maq output are dumped in compressed binary format. Human readable information are only extracted when necessary. Maq integrates most of functions in one executable, which is invoked as:
maq command [options] parameters [...]Commands will be introduced in the following sections. For more descriptions, see also the maq reference manual.
fasta2bfa: Convert Reference Sequences
Given reference sequences in a fasta file ref.fasta, command fasta2bfa converts the file into the BFA format:maq fasta2bfa ref.fasta ref.bfaLike all the binary files generated from maq, the output ref.bfa is endianness sensitive. This means, for example, the binary file generated in x86/Linux cannot be read in powerpc/AIX.
fastq2bfq: Convert Reads
Before doing the alignment, maq needs to convert read sequences into the BFQ. Given reads in a fastq file reads.fastq, command fastq2bfq achieves this function:
maq fastq2bfq reads.fastq reads.bfqThe output reads.bfq is compressed by zlib.
match: Align Reads to the Reference
The match command aligns reads in the BFQ format to the reference in the BFA format. If only one read file reads.bfq is provided, match command will do single-end alignment:
maq match out.map ref.bfa reads.bfqIf two read files, reads1.bfq and reads2.bfq for example, are provided, paired-end alignment will be performed:
maq match out.map ref.bfa reads1.bfq reads2.bfqAnd in this paired-end case, reads1.bfq and reads2.bfq must consist of the same number of reads, and the name of the n-th sequence in the first file must be identical to the n-th one in the second file. Minimum or maximum insert size can be adjusted on the command line by providing -i or -a options, respectively. More options are presented in the reference manual.
Maq usually performs better if the number of reads is around 2 million, which is about the number of reads in one lane of Solexa output. Too few or too many reads will likely yield bad efficiency.
The output out.map is a compressed binary file. It stores the sequences, qualities, positions of the mapped reads and related information such as the number of mismatches and the mapping quality. The positions are sorted to faciliate subsequent steps.
mapview: View the Read Alignment
The read sequences together with the positions can be extracted from out.map with command mapview:
maq mapview out.mapThe information will be printed on the screen, delimited by TAB. The format is explained in the maq reference manual.
mapmerge: Merge Read Alignments
As described above, match usually takes 2 million reads as input. What about large scale resequencing where 100 million reads will be generated? The preferred way is to divide reads into 50 batches with each batch containing about 2 million reads. After the alignment of 50 batches, we can merge the results with the mapmerge command. This command merge several alignments together and output one alignment in the same format as the input. For example:
maq mapmerge out.map in1.map in2.map in3.mapwhere out.map is the output.
In theory, mapmerge can merge unlimited number of alignments. However, as mapmerge will be reading all the inputs at the same time, it may hit the OS limit of the maximum number of opening files. At present, this has to be manually solved by endusers.
assemble: Build Mapping Assemblies
The ultimate goal of maq is to give reliable SNPs. Command assemble achieve this by taking the alignment file as the input:
maq assemble out.cns ref.bfa in.mapSome basic statistics will be output in the error output. Consensus sequences and qualities will be stored in out.cns, a compressed binary file. We will provide a Perl script to extract detailed information from this file.
Other Commands: Extract Information from Binary Files
Command cns2ref extract the reference sequences from the consensus file and cns2fq extract the consensus sequences and their qualities.
Command cns2snp directly gives the list of SNPs and additional information at the SNP sites. The format is explained in the maq reference manual.
Processing SOLiD Data
Maq has preliminary functions to process SOLiD data. See this page for more information.